当前聚焦:质谱在抗体分析中的应用
2023-06-25 11:31:52 来源: 弗戈工业在线
抗体类药物的研发日益增多,为了确保这些分子质量属性的准确描述,需要采用复杂的技术手段。质谱(MS)由于其优于目前现有分析技术的结构分辨能力,已经成为治疗性抗体研发的几乎必备的分析工具。质谱广泛应用于抗体研发的各个阶段,包括克隆选择、细胞培养过程开发、纯化过程开发、制剂开发、稳定性研究和可比性研究。质谱确定的结构特征包括氨基酸序列、二硫键位置、碳水化合物结构和特征以及许多不同的翻译后、过程中和贮存中的修饰。
在本综述中,我们将讨论用于单克隆抗体(mAbs)结构表征的各种基于质谱的技术。在讨论mAbs结构表征策略之前,有必要定义本文中使用的一些术语。通过质谱表征蛋白质的结构变异或修饰时,完整分子的质量测量可以呈现出整个蛋白质的一般特征,但不能提供变异或修饰的位置。换句话说,完整分子的质量测量并不能提供任何结构分辨信息。为了获得结构分辨的信息,需要在进行质量分析之前将蛋白质切割成较小的片段。这种切割过程可以在溶液中或气相中进行。当片段主要在溶液中生成时,质谱仪用于分析每个片段,分子根据从这些片段获得的信息构建:我们称这种方法为“up”类型方法。另一方面,当片段主要在质谱仪内的气相中生成时,质谱仪用于直接分析整个分子,我们称这种方法为“down”类型方法。
“up”和“down”方法进一步分为“bottom-up”、“middle-up”、“top-down”和“middle-down”方法亚型。方法亚型取决于引入质谱仪的分子大小相对于完整分子的大小。当直接将感兴趣的完整分子引入质谱仪时,该方法是“top-down”的方法。当蛋白质在引入质谱仪之前被切割成几个大片段时,如果直接测定这些片段的质量,则该方法是“middle-up”的方法;如果在质谱仪内进行进一步的片段化,则为“中间向下”的方法。当蛋白质在引入质谱仪之前被消化成小肽时,该方法是“middle-down”的方法。当蛋白质在引入质谱仪之前被消化成小肽时,该方法称为“bottom-up”方法,无论这些肽是否进行MS/MS。这些技术在结构分辨率、序列覆盖率、样品消耗和分析便利性方面各有优缺点。将这些方法应用于mAbs结构表征的总结如图1所示,将在以下章节中详细讨论。
 (相关资料图)
(相关资料图)
图1: 不同的基于质谱的技术用于单克隆抗体的结构表征
02基本概念2.1仪器分辨率
质量测量的质量取决于质谱仪的分辨率和质量准确性。高分辨率的仪器可以分辨质量稍有不同的分子,使它们的质量能够分别确定,而不是作为加权平均值合在一起。所有天然产生的蛋白质都具有普遍的质量异质性,这来自不同分子的不同同位素组成以及复杂发翻译后修饰。如果质谱仪具有足够的分辨率,那么这些同位素峰可以分辨,每个个体同位素组成的质量可以确定。在这些同位素峰中,单一同位素峰是最重要的,因为它是唯一具有独特同位素组成的峰。然而,对于如mAb这样的大型蛋白质,单一同位素峰是不可观察的,同位素分布如此广泛,以至于难以确定每个同位素峰中包含多少重稳定同位素。因此,同位素峰的精确质量用途有限。此外,对于像mAb这样大的蛋白质,同位素峰很难分辨。由于仪器分辨率与仪器灵敏度之间通常存在折衷,因此评估质量测定完整mAb的最佳分辨率是有帮助的。图2显示了一个典型的mAb(元素组成为C6600H10172N1738O2103S52)的模拟同位素分布作为分辨率(R=M/DM)的函数。可以看出,当分辨率在8000以上时,同位素包络峰宽度没有显著改变。尽管当分辨率达到200000时,同位素峰开始分辨,但它们的精确质量提供的信息很少,而且在这种高分辨率下,灵敏度会受到影响。因此,最佳分辨率约为8000-10000。分辨率在10000以上通常不会有助于分辨任何mAb异质性。在自然峰宽(半高处)达到25 Da的情况下,很难分辨出质量变化较小的异质性,如氧化(+16 Da),而用任何仪器几乎不可能分辨出脱酰胺(+1 Da
图2:典型单克隆抗体元素组成为C6600H10172N1738O2103S52,不同分辨率(R=M/ΔM)的模拟同位素分布。
2.2 质量准确度在一个分子的许多同位素峰中,单一同位素峰是唯一具有单一同位素组成的峰,因此具有唯一确定的理论质量。因此,在评估较小分子质量测定的准确性时,通常只使用单一同位素峰。然而,对于像mAb这样的大分子,同位素峰极难分辨。即使同位素峰被分辨出来,由于单一同位素峰的丰度几乎为零,无法可靠地确定单一同位素质量。因此,必须使用抗体的平均质量来确认其元素组成。事实上分子的平均质量受到组成该分子的元素的同位素丰度的影响。例如,在重组mAb的生产过程中,哺乳动物(CHO)细胞被喂养20种氨基酸和一些其他营养物质,这将确定产出抗体中的同位素丰度。因为不同元素的同位素丰度主要取决于它们的来源,因此,应该使用不同来源的元素的原子量来计算不同来源化合物的平均质量。表1显示了蛋白质中元素的IUPAC标准原子量和范围。表1中还显示了根据有机物中这些元素的同位素丰度估计的原子量及其范围。在计算天然蛋白质的理论质量时,最好使用有机来源元素的原子量,因为它们是这些分子中元素的主要来源。例如,对于元素组成为C6600H10172N1738O2103S52的mAb,其理论质量根据IUPAC标准原子量计算为149181.15 Da。但是,当使用根据有机来源估计的原子量时,计算出的理论质量为149181.89 Da。这个差异(0.74 Da)相当于mAb的理论平均质量差异为5.0 ppm。由于大自然中同位素丰度的变化,蛋白质的平均质量很容易因为几ppm而发生变化。例如,碳的原子量变化0.00008 Da(C3代谢过程的陆生植物范围的一半)会导致相同的mAb分子质量差异为0.53 Da(3.5 ppm)。类似地,其他元素的变化也会导致分子平均质量的低ppm差异(氢为1.6 ppm,氮为1.7 ppm,氧为1.3 ppm,硫为1.0 ppm)。总体而言,由于天然同位素丰度的变化,mAb的平均质量变化范围在3-5 ppm左右,前提是使用表1右列的有机来源元素的平均原子量。表1:常见原子不同来源的质量数因此,测量mAb质量的关键因素包括仪器分辨率和质量准确性。对于大型蛋白质如mAb,最佳分辨率约为8000-10000,分辨率在10000以上通常不会有助于分辨任何mAb异质性。质量准确度对于识别不同修饰和异质性也至关重要。理想情况下,蛋白质质量的测定应具有足够高的准确性,接近于天然同位素丰度变化所致的平均质量变化,从而有助于准确评估蛋白质的组成和可能的修饰.
近年来,飞行时间质谱(TOF)已在质量准确性(2-10 ppm)、分辨率(5,000-20,000)和最大m/z(高达10,000)方面取得了显著成果。当分析仪配备ESI时,这些特点非常适合测定完整mAbs的质量。
早期尝试使用ESI-TOF进行完整mAb质量分析的研究受到了当时仪器性能的限制。随着ESI和TOF技术的不断改进,ESI-TOF和ESI-Q-TOF已被广泛认为是分析大分子蛋白质(如mAbs)质量的标准仪器。
在ESI-TOF仪器上,使用每日外部校准,测定完整mAbs质量的质量准确性通常低于100 ppm。通过仔细执行实验,可以将完整mAb的质量准确性测定为25-50 ppm。在实验条件下优化以减少加合物形成,实验前进行校准,并严格控制去卷积参数,部分实验室可以实现接近10 ppm的质量准确性。
TOF通常可提供的最高分辨率比Orbitrap 和FTICR MS 低几倍,尽管最近的多通道TOF 分析仪能够实现超高分辨率(在m/z 400 处,R≥100000)。同时,TOF的分辨率在MS 和MS/MS 模式下基本相同,这在形式上是对Orbitrap 分析仪的一个优势,因为Orbitrap 在MS/MS 模式下常常为了速度而牺牲分辨率。然而,在实际应用中,TOF在MS/MS 模式下的较高分辨率并不一定能够转化为更高的质量精度(这通常是最理想的分析参数),因为TOF 的传输效率受到限制。因此,与TOF 相比,在类似的实验中,Orbitrap质谱仪实际报告的真正阳性识别率可能更高。
峰值检测限由信号/噪声比确定,由于检测器的暗电流、漂移离子以及“化学”背景离子的存在,TOF的噪声会增加。这些非共价的复合物由分析物离子、溶剂和有时还有气态分子组成,其电荷通常来自于质子以外的碱金属和其他附加物。这些非共价复合物在每个m/z单位处产生宽而模糊的峰,其峰谷由类似来源的多带电离子和其亚稳解离产物填充。这种化学背景的存在通常是限制TOF仪器检测阈值和动态范围的主要因素。有点讽刺的是,使用FT分析器(包括FTICR和Orbitrap)获得的质谱图几乎不含有化学背景。这种现象的可能解释是,要被检测到(即使是在错误的m/z处),背景离子只需要进入TOF分析器,之后就可以由于碰撞而解离或偏离其路径,而这两个事件都不会显著影响检测概率。同时,在FT质谱图中产生一个尖锐的、软件识别的峰,需要离子在FT分析器内保持完整的、相干的运动状态与类似离子共存达到较长的时间,即多毫秒的时间。所有偏离轨道、亚稳的或不相干的离子要么不会被FT检测到,要么会贡献于宽阔、平滑的背景信号,这些信号可以很容易地被软件减去。因此,尽管TOF的灵敏度形式上更高,但FT分析器的真实检测极限可能是可比或更低的。
Orbitrap Orbitrap质谱是近期新增的分析工具之一,由Alexander Makarov发明。Orbitrap质谱仪由一个同心的中央电极和一个桶状电极组成,并在两个电极之间施加静电场。注入静电场的离子被两种力约束;静电场抵消离心力场,并使离子在中心电极上轴向和径向运动。轴向的谐振频率应用于m/z测量,因为这种振荡模式与离子能量无关。外部电极上的图像电流用于检测。
质量分辨率取决于采集时间(更具体地说是谐振次数),因此更高的质量分辨率需要较长的采集时间,可实现m/z=400处的200,000的质量分辨率,这比大多数TOF仪器高得多(PS:广告词而已,m/z=400在抗体分析中并不是一个典型的质荷比,分辨率随着m/z的上升会随之下降,可以见下图,单方面强调广告级别的分辨率并没有太大意义。PS的PS: Thermo打钱我就删,当然Orbitrap的性能包括易维护性确实比TOF好)。
Orbitrap与FTICR的分辨率与m/z的关系(所有数据均显示为0.76秒扫描)
商业上,Orbitrap质谱仪通过C-trap与线性离子阱相耦合。C-trap的作用是在进入Orbitrap质谱仪的入口处电动挤压离子以收敛。线性离子阱和Orbitrap质谱仪可以分别或组合使用。例如,在分析模式下,Orbitrap可以执行质量分析,而离子阱用于接口离子源,连续传输离子并定期将离子引入Orbitrap。两者可以同时操作,其中在Orbitrap中执行质量分析,而在线性离子阱中执行MS/MS或MSn碎片实验并进行质量分析。
03结构表征3.1Middle-UP的结构表征
虽然测定完整mAbs的质量可以给出蛋白质的整体表示,但它不能提供任何结构分辨率。要获得结构分辨信息,必须在质量分析之前将蛋白质切割成更小的片段。另一种方法,称为“Middle up”方法,包括在质谱分析之前将蛋白质(mAbs)切割成几个大片段。将mAb切割成几个大片段的一种方便方法是还原二硫键以生成重链和轻链。对单个链的质量分析可用于确认每个链的结构,或者定位每个单独链中的变体或修饰。
完全还原所有二硫键可以在变性条件下进行还原。在没有变性剂的情况下,可以选择性地还原链间二硫键,使链内二硫键保持完整。这种简单的还原方法已被用于确认mAb结构,表征抗体结构,确定轻链和重链上的不同修饰,并检查重链上的糖基化结构。
另一种常见的中间向上方法是在天然条件下用蛋白酶(如木瓜蛋白酶,胃蛋白酶)对mAbs进行处理。对于许多IgG类分子,这些有限消化的切割位点通常在铰链区附近,生成Fab、F(ab)2和Fc片段。然而,IgG2分子在天然条件下抵抗酶解。这些通过酶生成的片段还原后会产生更小的片段。这些片段的质量分析提供了比完整mAb质量测量更详细的结构信息。与还原方法类似,有限消化方法已被用于确认mAb结构,识别翻译后或化学修饰,以及表征抗体结构。
与完整的mAbs相比,这些片段要小得多,因此更容易分析,可能在不太理想的仪器上具有更高的质量准确度。例如,对于IgG1分子,重链质量约为51 kDa,轻链23 kDa,Fc 53 kDa,单链Fc 26 kDa,Fab重链(Fd)24 kDa,Fab 48 kDa,F(ab)2 96 kDa,相较之下,完整的IgG1质量为150 kDa。由于要分析的分子尺寸显著减小,而且大多数片段不受糖基化异质性影响,因此减少了质谱仪的质量范围和分辨率要求。因此,这些片段可以在大多数仪器上进行分析,如四极杆或离子阱,尽管ESI-TOF/Orbitrap仍然是首选方法。
通过有限消化和/或还原产生的片段通常通过RP-HPLC分离,并与ESI-MS在线分析。例如,将DTT还原或木瓜蛋白酶消化应用于IgG1分子,然后进行RP-HPLC/MS分析,以表征和定量诸如焦谷氨酸环化、脱酰胺化、天冬酸异构化和氧化等多种修饰。还原或消化后,RP-HPLC上的亚结构分离良好。图3显示了木瓜蛋白酶消化后的IgG1进行质谱分析的总离子色谱图。图中还展示了峰5-7的质谱峰。从确定的质量可以明显看出,峰5是具有不同糖基的Fc区域,峰6是氧化的Fc区域,峰7是具有去酰胺的Fc区域。峰1-4被确定为Fab区域的不同形式,包括焦谷氨酸环化和脱酰胺(未显示在图中)。
图3
Middle up分析提供了一种简单的方法,将结构变化定位到分子的特定区域。这种方法具有一定程度的结构分辨率,但无法将变化定位到特定的氨基酸残基。这种方法对于确定抗体中结构变化的位置非常有用,但对于确定结构变化发生在哪个具体氨基酸上,则需要更高分辨率的方法。
3.2 BOTTOM-UP的结构表征
要确认蛋白质序列或在残基水平上表征修饰,最常用的技术是bottom up的方法。在bottom up的方法中,蛋白质被蛋白酶消化成小肽,然后进行LC/MS,或更常见的LC/MS/MS分析。蛋白水解与LC/MS/MS分析相结合,提供了高结构分辨率,但在质谱分析中,它通常是最耗材料、耗时且劳动密集的。消化过程中的假阳性,包括易失修饰如琥珀酰亚胺的丢失,以及氨基酸重排,可能导致错误的结论,因此需谨慎处理。bottom up广泛用于确认蛋白质序列,表征翻译后、过程中和贮存中的修饰,以及表征二硫键连接。
A.蛋白水解通常在变性条件下对mAb进行还原和烷基化后,使用胰蛋白酶或Lys-C进行蛋白水解。其他不太常用的蛋白酶包括Glu-C(V8)和Asp-N。化学切割方法,如氰酸溴(CNBr)消化,也偶尔会使用。
以上所有方法通常将蛋白质切割成适当大小的肽,这些肽在MS/MS实验中能有效地片段。需要注意的是,在大多数商用仪器上,大肽(>4 kDa)很难通过MS/MS表征,而非常小的肽(2-3个残基)通常由于在反相柱上的保留不佳而丢失。
B. LC/MS和 LC/MS/MS 在早期,MALDI-TOF仪器广泛用于测定从mAb和其他蛋白质的蛋白水解产生的肽的质量,用于结构确认和异构体/修饰分析。与ESI-MS相比,MALDI-TOF具有易用性,且耗材和时间较少的优点。然而,由于LC串联的便利性和肽鉴定所需的串联质谱质量较好,目前大多数在mAb上进行的自下而上实验都是在ESI仪器上进行的。
在LC/MS/MS肽图实验中,困难通常出现在mAb的蛋白水解上。通常需要大量的蛋白质材料才能获得成功的消化。只要实现完全消化,由于现代仪器的显著进步,MS检测的灵敏度通常不是问题。因此,大多数LC/MS/MS肽图实验是在小孔径RP-UPLC柱上进行,以获得最佳的色谱分辨率。
C.二硫键确定要确定蛋白质内的二硫键连接方式,必须在二硫键仍然完整的情况下进行消化。在普通条件下,单克隆抗体分子非常稳定并且难以消化,因此单克隆抗体内的二硫键连接方式的表征具有挑战性。成功的消化关键在于在强性变性条件下使单克隆抗体变性,例如6M盐酸胍和高温。重要的是,在变性过程中必须存在烷基化试剂,如碘乙酸(IAA),碘乙酰胺(IAM)或N-乙基马来酰亚胺(NEM),阻止单克隆抗体中所有存在的自由巯基组来防止二硫键杂化。由于其在较低pH下的活性,通常优先使用NEM。消化前必须稀释变性剂以保留蛋白酶的活性。消化通常使用胰蛋白酶或Lys-C。随后,一部分样品会将使用DTT或者TCEP进一步还原二硫键,并将LC / MS / MS肽图与未还原图进行比较以确定二硫键连接。靠近铰链区域的二硫键通常使用Edman测序或者使用TCEP在弱酸性下在NEM or M-biotin存在的条件下部分还原检测。
另一个用于确定mAb中二硫键的有用方法是在负离子模式下通过LC/MS分析非还原消化物。在负离子模式下,二硫键通常被有效地断裂,以显示通过二硫键连接的链。这种方法可以被视为在气相中的二硫键还原。
治疗性抗体的二硫键结构通常必须经过确认,以确保抗体被正确制备和折叠。大多数重组的单克隆抗体确实显示出与预期结构一致的二硫键结构,除了IgG2分子。有证据表明,重组和自然发生的IgG2分子中存在二硫键的异质体。当在非还原条件下消化重组的IgG2分子时,观察到一些后期洗脱的RP-HPLC峰。这些峰的质谱分析以及这些峰的收集和还原形式的质谱分析表明,它们是二硫键连接的肽;所有这些肽都涉及连接到轻链恒定区和/或CH1域的铰链区。这个结果表明,在IgG2分子中存在二硫键变异体。这些二硫键变异体可以通过离子交换和反相色谱分离,并且它们在全血或“血液样”环境中相互转化。进一步的研究表明,这些二硫键变异形式也存在于人骨髓瘤IgG2和人血清多克隆抗体中。
3.3 TOP-DOWN结构表征
尽管bottom up的方法提供了最多的结构信息,但是它们需要大量的人力,而且常常存在一些问题,比如需要消耗大量样品,消化过程中可能会引入人为因素,以及样品制备需要花费很长时间。对于数量或浓度有限的样品,例如从色谱分离中获得的微量组分,需要进行多次收集和浓缩才能获得足够的材料进行成功的酶解。在这种情况下,top-down和middle-down成为提供有用序列信息的非常快速和方便的选择。
Top-down质谱法是指将完整分子引入质谱仪,而不进行溶液中的酶解或化学分解,并通过分析分子在质谱仪内的裂解模式来获得结构信息。自上而下的质谱法可成功地快速表征小到中等大小的蛋白质。由于产生的高度带电的碎片离子数量庞大,因此通常需要高分辨质谱仪进行top-down的质谱分析。通常来说,可以通过仪器分辨率来确定可用自上而下质谱法分析的蛋白质大小。例如,分辨率为10,000的Q-TOF型仪器应该可以为重量达到10,000 Da的蛋白质提供大量的结构信息。对于更大的蛋白质,首选具有更高分辨率的仪器,如傅里叶变换离子回旋共振质谱(FT-ICR MS)或Orbitrap
图4展示了当在Thermo-Fisher LTQ-Orbitrap上分析IgG2分子时的in-source fragmentation图谱。除了小的N末端轻链碎片和小的N末端和C末端重链碎片以及一些内部碎片离子外,一个相当引人注目的观察结果是存在一系列大离子,这些离子的存在表明在重链的b106到b118离子和轻链的b114到b118离子之间存在一些结构信息。这些N末端碎片对应于重链和轻链的整个可变区域。可以对这些b离子进行MS/MS以获得有关可变区域更详细的结构信息。
图4:一种IgG2分子in-source fragmentation质谱图。完整的图谱显示在顶部。底部显示了从m/z 1,500到1,850的图谱。主要的碎片离子用带电荷的b或y离子标记。前缀H代表重链,L代表轻链。Top-down的质谱法消耗的材料很少,可以在很短的时间内提供有用的结构信息。当与在线液相色谱/质谱法相结合时,in-source fragmentation的完整mAb可以在没有任何样品制备的情况下提供有用的可变区域以及末端区域的结构信息。
自上而下的质谱法用于大蛋白质(如mAb)的主要缺点是通常具有有限的结构分辨率。对于IgG1和IgG2分子,只能获得可变区域和末端附近的信息。在大多数情况下,机缘巧合仍然是决定实验结果的重要因素。如果感兴趣的修饰位点位于富含序列信息的区域,则可以准确地确定修饰位点。在其他情况下,如果修饰位点位于序列信息较少的区域,则可以将修饰位点分配给蛋白质的一个较大的片段,但无法指定特定的氨基酸残基。
3.4 MIDDLE-DOWN结构表征
Middle down可能是解决自top down中结构分辨率有限问题的一种有价值的替代方法。在middle down中,蛋白质首先通过还原二硫键或者有限的蛋白酶水解作为中间步骤分成几个大片段,然后再被引入质谱仪进行MS/MS分析。与top down相比,middle down可以在一些有限的样品制备情况下潜在地提高结构分辨率。由于中等质量实验中所采用的还原二硫键是化学反应,可以使用大量的化学试剂来实现,因此它不像酶反应那样有样品浓度的要求,因为酶反应通常不使用大量的酶(贵贵贵贵啊)。因此,对于稀释的少量样品,middle down实验比top down更有优势。
图5显示了一种IgG2分子的轻链和重链的CID MS/MS谱图,该谱图是在Thermo LTQ-Orbitrap上获得的。Orbitrap的超高分辨率和质量精度可确保片段离子的准确鉴定。通过MassAnalyzer的分配和手动验证,根据准确测定的质量、同位素模式和一些片段化规则,大多数片段离子可以分配为b、y或其他片段。根据这些分配,轻链中213个肽键中有59个被切割,重链中447个肽键中有67个被切割;这些数据对应于轻链的平均结构分辨率(通过将肽键的总数除以被切割的肽键数来计算两个切割位点之间的平均残基数)为3.6个残基,重链为6.7个残基。
图5
04未来展望直到最近,几乎所有的mAb结构表征都是通过bottom-up方法实现的。虽然bottom-up方法越来越成熟,但是top-down和middle-down方法仍处于发展初期。然而,top-down/middle-down方法的优点,如最小样品操作和快速反应时间,使得这些技术非常有吸引力。虽然并不是所有的结构问题都能用top-down或middle-down方法解决,但许多问题可以在几个小时内解决,而不是像"up"方法一样需要几天时间。top-down/middle-down方法没有被广泛接受的主要原因之一是它们需要高分辨率的仪器。然而,现代高分辨率仪器,如FT-ICR和Orbitrap,正在变得更加用户友好,并且这些高端仪器正逐渐放到工业MS用户手中。 当表征蛋白质异构体或修饰时,"bottom-up"方法将逐渐成为辅助方法,只有在"top-down"或"middle-down"实验失败时才会使用。电子俘获解离(ECD)在蛋白质结构分析的"up"和"down"方法中具有巨大潜力。然而,由于使用难度大,需要FT-ICR仪器,因此它并没有成为蛋白质化学家的主要工具。然而,最近开发的电子转移解离(ETD)技术被广泛应用于离子阱仪器中。此外,在Orbitrap上实现ETD使得对更大的肽段进行结构分析成为可能,并且对于middle-down实验来说成为了理想的选择。参考文献:Roman A. Zubarev and Alexander Makarov.Orbitrap Mass Spectrometry. doi.org/10.1021/ac4001223
Zhongqi Zhang, Hai Pan, and Xiaoyu Chen. Mass spectrometry for structural characterization of therapeutic antibodies. DOI 10.1002/mas.20190


热点推荐
-

工龄与退休后工资领取对照表 工龄30年能领多少退休金?
-
嘉凯城(000918.SZ)今日起临时停牌 复牌时间未知
-
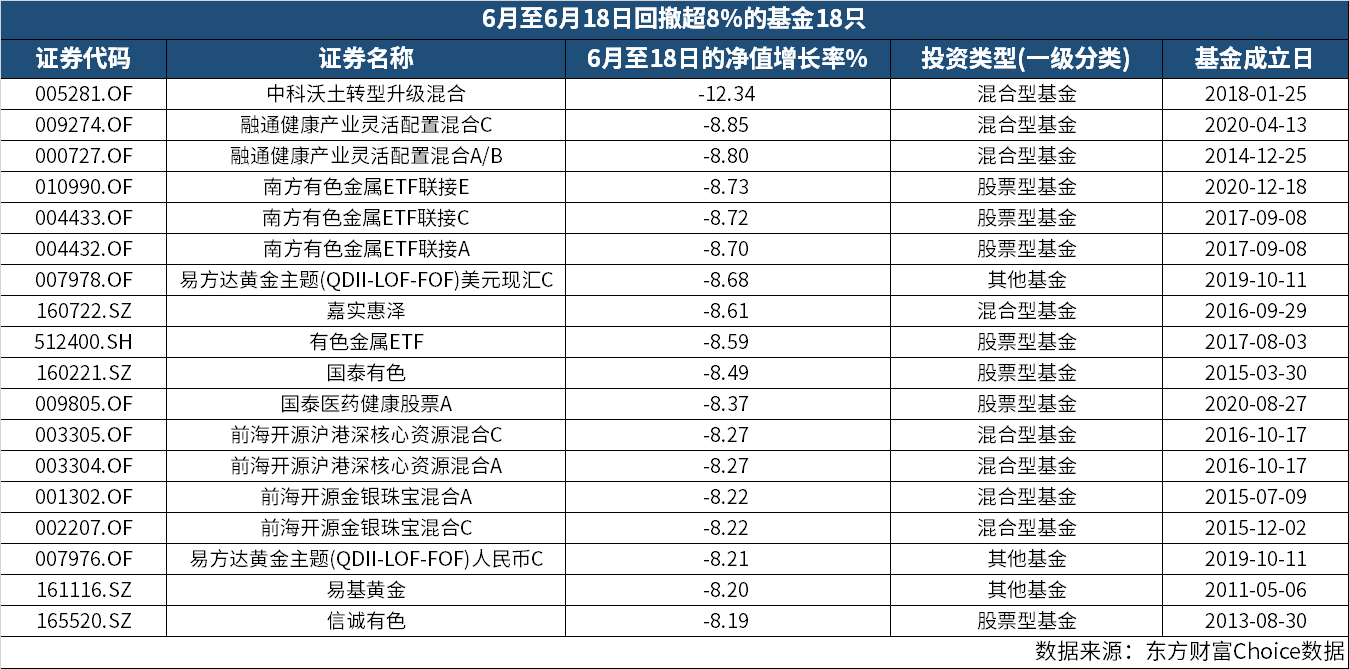
中科沃土转型升级混合 6月回撤已超12%
-
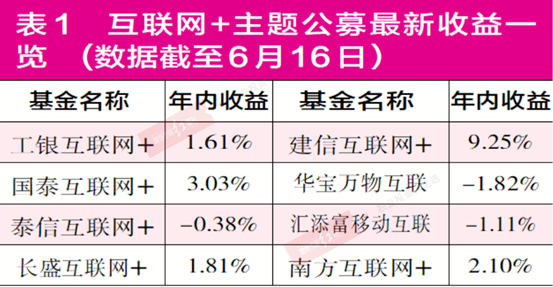
“互联网+”产品普遍举步维艰 多家产品被“深套”
-
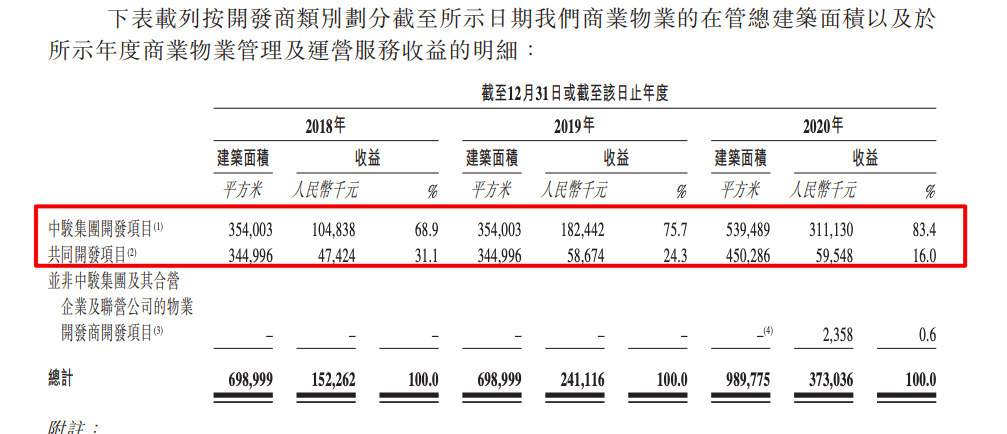
中骏商管将挂牌 中骏控股集团持股62.43%
-
当前聚焦:质谱在抗体分析中的应用
-

2023第十五届“八喜杯”小篮球精英赛落幕
-

课后服务行业市场相关政策 课后服务行业市场发展策略研究 最新
-

靶向治理领导干部违规插手等问题 斩断工程建设领域腐败利益链_快资讯
-

持续高温天气成因有哪些?何时结束?北方为何热过南方?_世界新动态
-

制作镜像系统u盘安装系统-(制作镜像系统u盘安装系统教程)
-

烟台市福山区第二实验幼儿园幼小衔接之参观小学实践活动
-

实景倾呈 品质共鉴丨华昌·金沙一品交出惊艳答卷 全球热门
-

世界速递!陵水养老哪个地方好?东北人在海南陵水县买房主要养老买那个位置好?
-

高考查分的心情你还记得吗?一起沉浸式体验-每日观点
-

浮法玻璃龙头预计上半年净利下滑约四成!下周超千亿解禁洪流来袭,4股解禁超百亿
-

焦点快报!“强拆长城景区大门”:涉事董事长所属公司曾列为老赖被“限高”
-

环球快消息!特斯拉和华为押注4D成像雷达技术,谁家芯片方案最有看点?-天天即时看
-

她越裹越紧,在座各位都有责任-报道
-

海优新材:融资余额3.5亿元,创历史新高(06-08)
-

6月26日—28日,东莞虎门这些路段,将适时实施禁鸣喇叭
-
如何编蝴蝶结?
-

全球焦点!小于等于的数学符号_小于等于号符号怎么写
-

王者荣耀亚连怎么出装?王者荣耀亚连出装推荐|全球球精选
-
南非世界杯决赛中西班牙队的首粒进球是谁打进的_南非世界杯决赛
-
天天滚动:国风市集、亲子读书会、健康义诊……金海街道端午佳节精彩纷呈
-

全球聚焦:广州电信与广东省中医院签署战略合作协议
-

当前简讯:九江学院:用中国传统文化讲好中国故事
-
环球观点:四川省2023年普通高校对口招生和藏彝文一类模式高考录取控制分数线出炉—中国新闻网·四川新闻
-

当前快播:点赞!开颅3次的少年高考过二本线